Gene knockouts by exon deletion with Cas9 plasmid transfection
Gene knockouts by exon deletion with Cas9 plasmid transfection
Cas9 is a dream biotechnology, since you can use it to quite easily and specifically disrupt almost any DNA sequence you’re interested in modifying. Its been particularly helpful in work with cultured mammalian cells. In this document, I will describe how I use Cas9 to make gene knockouts in HEK 293T cells.
So although I’ve heard good things about transfecting guide-RNA loaded Cas9 ribonucleotide protein complexes into cells, I haven’t done this yet. Instead, I’ll describe a likely less optimal, but super cheap and easy method of co-transfecting Cas9 and guide-RNA expressing plasmids into cells to create knockouts. Specifically, I’ll describe my method of knocking out a gene by removing a big chunk of coding sequence, either ablating its start codon, or getting rid of an important exon to only create truncated or frameshifted non-functional proteins.
Equipment & Materials:
1) Tissue culture hood & incubator
2) Transfection reagent
3) Fluorescence Activated Cell sorter (FACS)
4) Plasmids from Addgene, IDT (or similar) account for oligo synthesis
5) PCR machine, polymerase, Gibson master mix.
6) Agarose gel electrophoresis apparatus and some form of GelDoc.
7) Sanger sequencer (or sequencing service)
Step1: Make custom guide-RNAs for your gene of interest
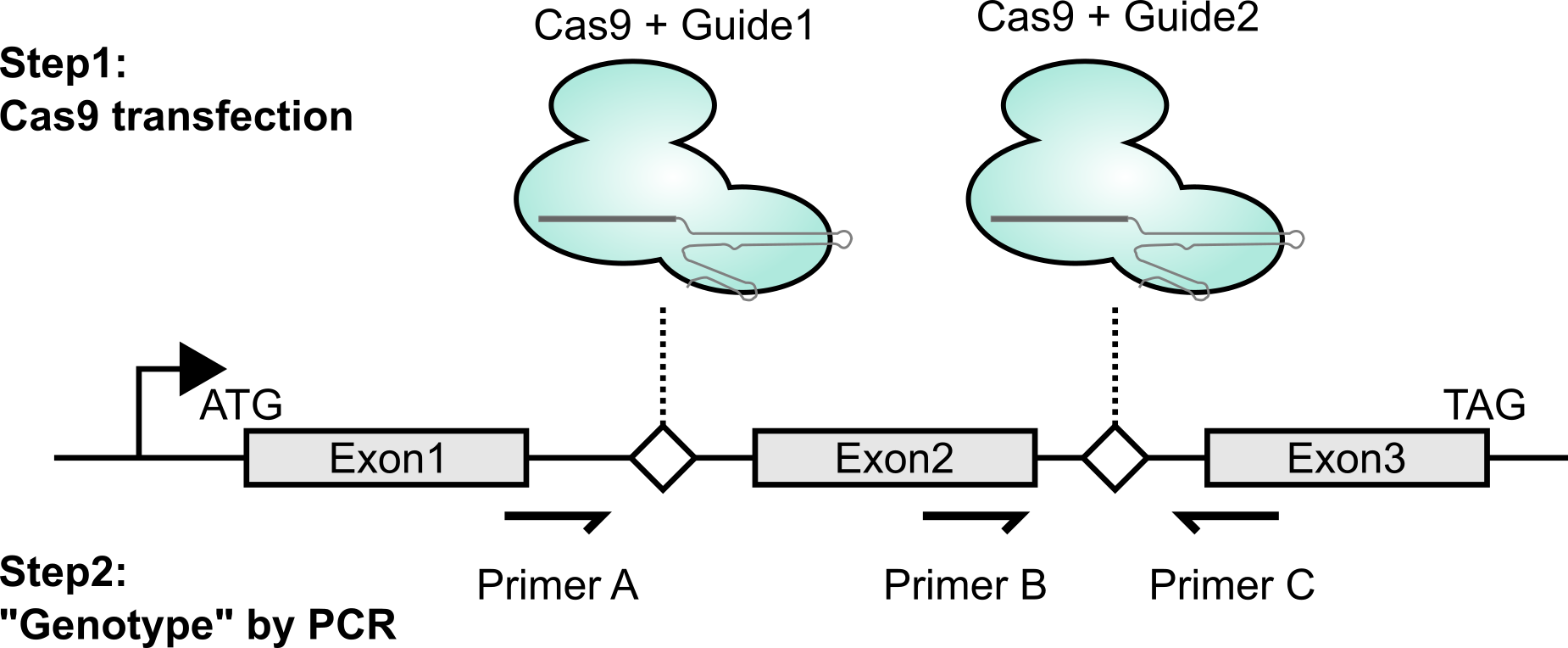
As mentioned above (and shown in the figure), to create genetic knockouts, I like to delete out entire exons. The main reason is that it’s really easy to genotype with PCR. It’s also nice in that if the change is made, even if the repair is an in-frame indel, the deletion will be so large that the protein should still be loss-of-function. Lastly, exon deletion can often be achieved by targeting adjacent introns; this means that even if you stably express the Cas9 / gRNA combo, you can re-express a cDNA version and have it not be susceptible to disruption.
I first find guide RNAs that will target the DNA I want to disrupt. Addgene seems to have a pretty handy list of guide-RNA sequence design applications here. By default, I still tend to go to this classic website. Have it come up with a pair of seemingly specific guides, per the strategy above. Note: When using a U6 Pol3 promoter, I’ve read that if your guide RNA doesn’t start with a G, you should plan to include an extra G in front of the guide sequence to increase transcription.
Addgene lists a bunch of protocols from the original labs here. Depending on how you like to do your molecular cloning, you may want to follow one of those. I’ll describe making a custom guide-RNA using Gibson Assembly, which is absolutely amazing.
Using a really basic U6-promoter guide-RNA expressing plasmid (like this), design a pair of long primers that overlap in their 5′ ends with the guide-RNA sequence, but hybridize to sequences adjacent to the guide-RNA sequence but point in the opposite directions. I shoot for a primer hybridizing sequence with a melting temp at or approaching 60*C. The ~20nt guide-RNA sequence makes for a great Gibson overhang.

The steps here are:
1) Amplify plasmid, appending the guide-RNA sequence to the termini of the amplicon. I use Kapa Hifi, with 40ng template plasmid (30ul total volume), and ~7/8 cycles.
2) DPNI digest. I pipet 1ul of DPNI directly into the 30ul reaction tube, and incubate at 37*C for 2 hours (I suspect you don’t need all two hours. DPNI is supposed to work sub-optimally in Kapa Hifi buffer, but works well enough with these conditions).
3) “Purify” your intended plasmid. At first, I used to run my amplifications out on an agarose gel and gel extract (using a Qiagen gel extract kit). Nowdays, I just use a Zymo Clean and Concentrate kit (first setting aside some sample to run on a gel afterwards, for diagnostic purposes mostly). I elute in a small volume (6 ul for the Zymo kit).
4) I then make equal volume mixtures (eg. 1ul “insert”, 1ul “vector”), and mix in the corresponding amount of 2x Gibson Master Mix (So 2ul for the above example). I incubate this at 50*C for 0.5 to 1 hr. Standard protocol here.
5) I mix the sample (usually 4ul) into 50ul of chemically competent E.coli, and transform (eg. “Heatshock” transformation).
6) In the case of the Mali et al Church lab gRNA plasmids, they’re Kan resistant, so don’t forget to recover before plating onto Kan plates.
Normally, I end up seeing tens to hundreds of colonies the next day. I then usually pick 2 colonies to grow up and miniprep. When I run Sanger, usually at least one colony contains plasmid DNA with the intended guide-RNA encoded. If neither colony has the intended plasmid, I normally pick two more and screen those. I don’t remember the last time this protocol failed (possibly hasn’t in a dozen or so attempts), but I think if it does it’s likely going to be from there being too much template, so I would then re-do the process really making sure DPNI is doing its job (and maybe gel extracting if I have to).
Step2: Transfecting the guide-RNA and growing clonal lines
I’ve only ever done this in HEK 293T cells, since that’s where I do most of my tissue culture work (since they’re so easy to handle). Next step is to transfect cells with the guide-RNA and Cas9 encoding plasmids. I tend to do this in 6-wells, using Fugene 6.
For this transfection, I either mix the guide-RNA plasmids (Equal amounts of plasmids with guide 1 and guide 2) with PX458 (which encodes Cas9 2A-linked with EGFP), or a plasmid expressing untagged Cas9 along with a plasmid encoding a fluorescent protein. Since transfection usually results in many many copies (hundreds? thousands?) of plasmid getting in per cell, cells expressing the fluorescent protein (generally) should have been transfected with guide RNAs and Cas9 also (and thus are the cells most likely to have been edited).
Then, approximately two days after transfection, I sort for transfected cells using the fluorescent marker. I usually do this in a 96-well plate, sorting single cells into individual wells (to create clonal lines), while taking one of the corner wells and sorting 100+ cells into it. This gives me an improved bulk population as backup in case the single clone growouts fail (Generally 1/4 to 1/3 of all sorted wells grow out using HEK 293T cells for me), and gives me a well for focusing onto the right focal plane using the light microscope (since it’s hard to correctly focus on wells where it’s not clear if there are even any cells there).
If in a rush, I’ll check on the wells in approximately a week. By then, you can definitely see colonies of cells growing by brightfield microscope. I would then trypsinize and replate to keep them growing happily. If I’m not in a rush, I’ll sometimes let the cells grow for a couple weeks overall before checking; at this point, the media color starts becoming yellowish in wells containing growing cells, and clonal lines that grew out are large enough that if I transfer them to a new well, I’ll be able to do an expt with those cells in a few days.
Step3: PCR Genotyping
Once I have enough cells (say, at least half a 24-well plate well’s worth), I’ll extract genomic DNA using a Qiagen DNA easy kit. I’ll then use the primer combinations shown above in the first figure. Notably, primer combo BC should only amplify if there is a wild-type copy present (DNA from unmanipulated cells should serve as a positive control). This should be a binary “amplifies or doesn’t” readout. The other primer combo also gives a slightly orthogonal perspective on what might be present at the intended locus within the genome. If unmodified, combo AC should give a large band (or no band, if these are really large introns). If modified, combo AC should give a much smaller product (I aim for a few hundred bases for easy visualization by agarose gel electrophoresis). To verify that this small band is what I think it is, I gel extract the band and sequence using Sanger. Conveniently, you can even use either primer A or primer B as the Sanger sequencing primer if you didn’t already order a dedicated internal primer. In the handful of times I’ve done this, I think the predominant modified product that I’ve seen is literally a blunt-ended Non-Homologous End Joining of the two DNA fragments without any removal of terminal nucleotides. Thus, if this is indeed the major modified product, I’m able to design my guides such that such NHEJ ligation results in a frameshift.
Using the above PCR, I go through and screen the colonies to find one that looks to be a complete knockout (no amplification with primers BC, only small band with primers AC). In the handful of times that I’ve done it, screening a dozen colonies has yielded multiple knockout clones. Of course, actual protein knockout should be confirmed by western blot.
Voila! Knockout cells to now use in your experiments! But, as I noted previously, I’ve only done this a few times (albeit pretty successfully each time), so in no way do I guarantee that it will absolutely work for you the first time.
Some references not already cited:
I think this project / paper from super grad student Molly Gasperini in Jay Shendure’s lab is what originally made me think about using paired gRNA deletions to make knockouts.
I’ve tried to link to specific protocols in the above descriptions. For any details that are ambiguous, the methods described in my 2017 NAR paper should largely describe the basic steps / reagents used (though to be clear: I did not actually use this knockout method in this paper).